La policitemia es un trastorno en el cual hay demasiados glóbulos rojos en la circulación sanguínea. Es el opuesto de la anemia, que ocurre cuando hay escasez de glóbulos rojos en la circulación. También se denomina plétora (aumento excesivo de sangre).Esto puede ocurrir si a una dada presión de oxígeno la Hb queda saturada de O2 no liberándolo como las Hb normales en las mismas condiciones. El organismo para compensar origina más glóbulos rojos. Sus causa pueden ser las siguientes:
aumento en la producción de glóbulos rojos:

*Un feto con niveles de oxígeno reducidos crónicamente responde produciendo glóbulos rojos adicionales .Algunas anomalías cromosómicas pueden provocar mayor producción de glóbulos rojos.
*Los glóbulos rojos adicionales ingresan a la circulación del bebé de otra fuente:
*Un retraso en la sujeción del cordón umbilical con abrazaderas luego del parto hace que sangre de la placenta ingrese en la circulación del bebé.
Aquellos nacidos a grandes altitudes, debido a la mayor demanda de sangre para transportar oxígeno.
*aquellos nacidos luego de 42 semanas de gestación.
Los pequeños para la edad gestacional (SGA) / los que padecen retardo de crecimiento intrauterino (RCIU).
*los gemelos que comparten la placenta y desarrollan el síndrome de los gemelos transfusor-transfundido.
*Los bebés de madres diabéticas.
aquellos con anomalías cromosómicas, incluyendo trisomías 13, 18 y 21 (síndrome de Down).
Síntomas
Muchos bebés con policitemia no tienen síntomas visibles del trastorno. A continuación se enumeran los síntomas más comunes de la policitemia. Sin embargo, cada bebé puede experimentarlos de una forma diferente. Los síntomas pueden incluir:
*coloración rojiza-púrpura profunda
*mala alimentación
*letargo
*bajo nivel de glucosa en sangre
*A veces dolor de cabeza por congestión de vasos.
*Rotura de vasos.
*Trastornos de circulación.
*Trombosis.
*A veces dolor de cabeza por congestión de vasos.
*Rotura de vasos.
*Trastornos de circulación.
*Trombosis.

Diagnóstico
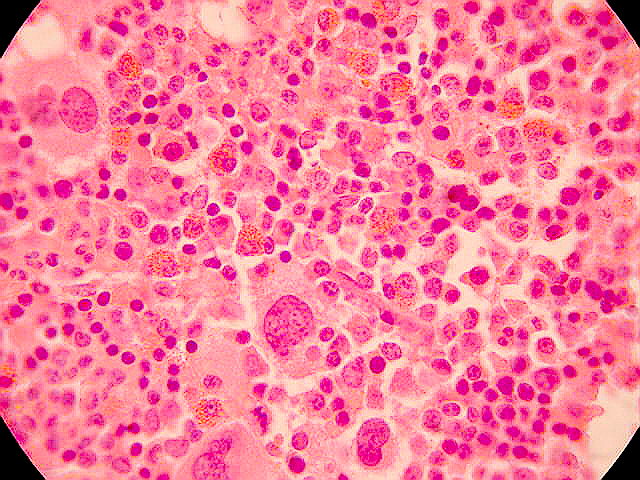
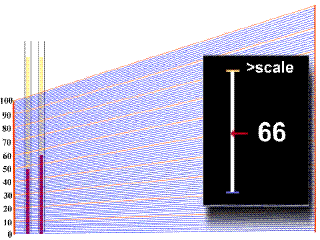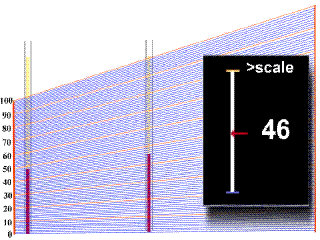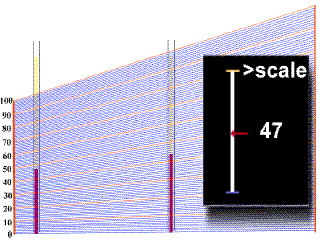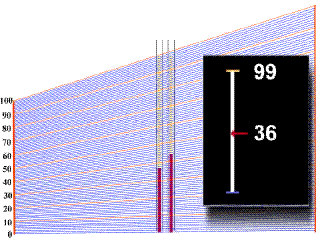
Las pruebas de laboratorio muestran un hematócrito alto (recuento de glóbulos rojos) cuando existe policitemia. Un nivel de hemoglobina alto (proteína de la sangre que transporta oxígeno) también puede ayudar a diagnosticar la policitemia.
Tipos
Policitemia se refiere al aumento del número de hematíes y, generalmente, de la cantidad de hemoglobina. Hay dos tipos:
Policitemia esencial o Policitemia vera: Es de causa desconocida, aunque muchas veces está en el contexto de un síndrome mieloproliferativo.
Policitemia secundaria a:
- Algunos tipos de tumores.
- Endocrina: en el síndrome de Cushing
El tratamiento
puede incluir:
Extraer una parte del volumen de sangre, reduciendo así la cantidad de glóbulos rojos.
Reemplazar la sangre extraída con líquidos (para ayudar a diluir la concentración de glóbulos rojos).
Exsanguíneo transfusión parcial (extraer y reemplazar lentamente una gran parte del volumen de sangre del bebé).










{kind=link}