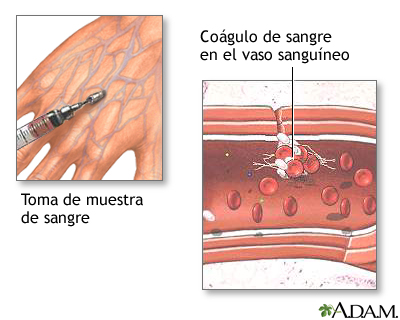
Este examen mide la cantidad de tiempo que su sangre toma para coagular. Este examen puede ser usado para supervisar el tiempo de coagulación cuando heparina (un medicamento de aclaración de sangre) es usada.

TIEMPO PARCIAL DE TOMBOPLASTINA: Es una prueba de sangre que examina el tiempo que le toma a la sangre coagularse y puede ayudar a establecer si uno tiene problemas de sangrado o de coagulación.

DETERMINACIÓN DE FIBRINÓGENO: La determinación de fibrinógeno puede llevarse a cabo junto a otras pruebas de factores de la coagulación cuando se sospecha que el paciente puede tener una disfunción o un déficit hereditario de un factor, o cuando el médico necesite evaluar y monitorizar la capacidad de coagulación (a lo largo del tiempo) de un paciente con un trastorno de sangrado adquirido.
A veces, la determinación de fibrinógeno se realiza junto a otras pruebas cuando el médico pretende evaluar el riesgo de un paciente de desarrollar enfermedad cardiovascular.

EL TIEMPO DE LA COAGULACIÓN: Determina el tiempo que tarda en coagular la sangre recién extraída. Evalúa la vía intrínseca de la coagulación. Al mismo tiempo evalúa en términos generales: el fibrinógeno y el número y calidad de las plaquetas. Sirve además para controlar los tratamientos con heparina aunque con menos certeza que el tiempo parcial de tromboplastina activada.

REALIZA LA VALORACION DE:
Factor vascular. Factor plaquetario
VALOR NORMAL
Hasta 4 minutos

TIEMPO DE COAGULACION (Lee-White)
itativa del sistema de coagulación.Es usado para determinar el tiempo de retraccionde coagulo.
Factor plaquetario. Todos los factores plasmáticos de la coagulación.
Hasta 12 minutos






















{kind=link}
{kind=link}
{kind=link}